Just five years ago, the world of variant interpretation was very different. There was no standard terminology for variant classification, and laboratories were using many different terms as ambiguous as “mutation” and “polymorphism” to convey pathogenicity, or lack thereof, and a plethora of qualifiers such as “possibly,” “probably,” and “likely” to convey degrees of uncertainty. The only terminology broadly agreed on was “variant of uncertain significance” (VUS). Furthermore, there was substantial variability in how laboratories evaluated evidence. A single publication in a peer-reviewed journal could be cited as justification for declaring pathogenicity, even though on many occasions little evidence to support that assertion was present in the publication beyond, for example, a non-statistically-significant observation in an affected individual. This was perpetuated by the routine deposition of “deleterious mutations” in the Human Gene Mutation Database,1 often without strong evidence supporting these claims. Similarly, exemplar variants included in the Online Mendelian Inheritance in Man database2 to support gene–disease relationships were assumed to be pathogenic.
Dr. Khera is a physician-scientist with expertise in epidemiology, clinical medicine, and human genetics. Among his scientific contributions, he pioneered a new approach to quantify genetic risk for common diseases, […]
Ben Kleinstiver is a biochemist and genome editor whose interests include translating technologies into molecular medicines. He received his Ph.D in Biochemistry from the University of Western Ontario, and completed […]
Amit V. Khera, MD, MSc Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations
Nature Genetics. volume 50, pages1219–1224 (2018)
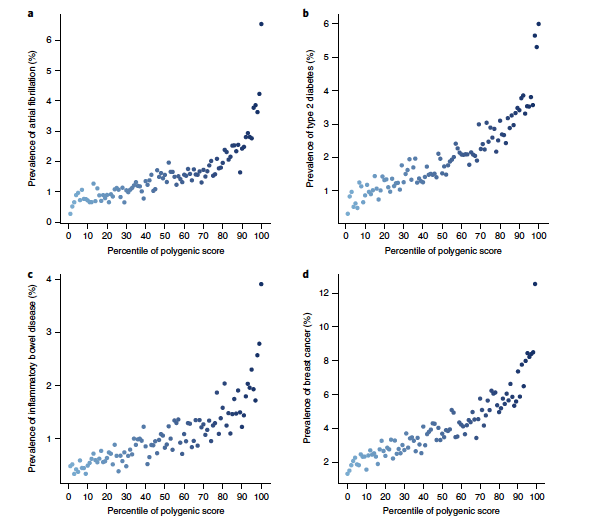
A key public health need is to identify individuals at high risk for a given disease to enable enhanced screening or preventive therapies. Because most common diseases have a genetic component, one important approach is to stratify individuals based on inherited DNA variation1. Proposed clinical applications have largely focused on finding carriers of rare monogenic mutations at several-fold increased risk. Although most disease risk is polygenic in nature2,3,4,5, it has not yet been possible to use polygenic predictors to identify individuals at risk comparable to monogenic mutations. Here, we develop and validate genome-wide polygenic scores for five common diseases. The approach identifies 8.0, 6.1, 3.5, 3.2, and 1.5% of the population at greater than threefold increased risk for coronary artery disease, atrial fibrillation, type 2 diabetes, inflammatory bowel disease, and breast cancer, respectively. For coronary artery disease, this prevalence is 20-fold higher than the carrier frequency of rare monogenic mutations conferring comparable risk6. We propose that it is time to contemplate the inclusion of polygenic risk prediction in clinical care, and discuss relevant issues.
Heidi Rehm is the Chief Genomics Officer in the Department of Medicine and at the Center for Genomic Medicine, the Medical Director of the Broad Institute Clinical Research Sequencing Platform […]
Nat Genet. 2017 Jan;49(1):36-45. doi: 10.1038/ng.3720. Epub 2016 Nov 14.
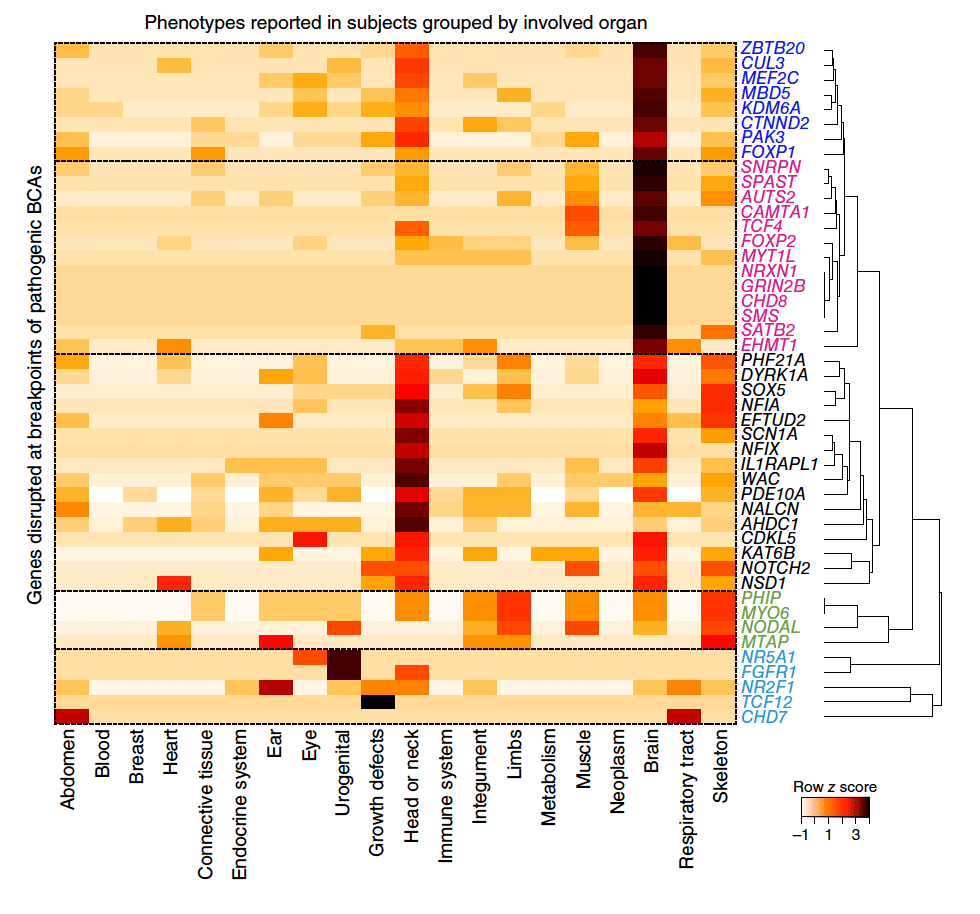
Despite the clinical significance of balanced chromosomal abnormalities (BCAs), their characterization has largely been restricted to cytogenetic resolution. We explored the landscape of BCAs at nucleotide resolution in 273 subjects with a spectrum of congenital anomalies. Whole-genome sequencing revised 93% of karyotypes and demonstrated complexity that was cryptic to karyotyping in 21% of BCAs, highlighting the limitations of conventional cytogenetic approaches. At least 33.9% of BCAs resulted in gene disruption that likely contributed to the developmental phenotype, 5.2% were associated with pathogenic genomic imbalances, and 7.3% disrupted topologically associated domains (TADs) encompassing known syndromic loci. Remarkably, BCA breakpoints in eight subjects altered a single TAD encompassing MEF2C, a known driver of 5q14.3 microdeletion syndrome, resulting in decreased MEF2C expression. We propose that sequence-level resolution dramatically improves prediction of clinical outcomes for balanced rearrangements and provides insight into new pathogenic mechanisms, such as altered regulation due to changes in chromosome topology.
Roy H. Perlis, MD, MSc Patient-specific models of microglia-mediated engulfment of synapses and neural progenitors
Mol Psychiatry. 2016 Dec 13. doi: 10.1038/mp.2016.220. [Epub ahead of print]
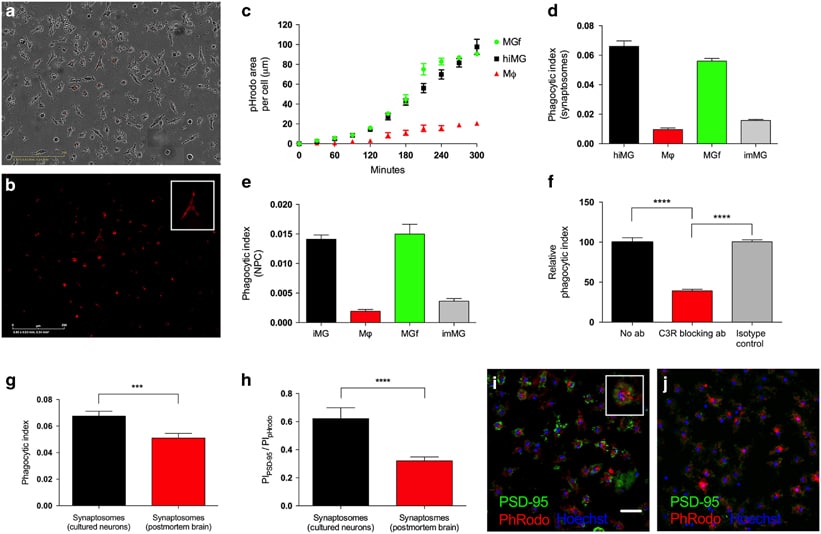
Engulfment of synapses and neural progenitor cells (NPCs) by microglia is critical for the development and maintenance of proper brain circuitry, and has been implicated in neurodevelopmental as well as neurodegenerative disease etiology. We have developed and validated models of these mechanisms by reprogramming microglia-like cells from peripheral blood mononuclear cells, and combining them with NPCs and neurons derived from induced pluripotent stem cells to create patient-specific cellular models of complement-dependent synaptic pruning and elimination of NPCs. The resulting microglia-like cells express appropriate markers and function as primary human microglia, while patient-matched macrophages differ markedly. As a demonstration of disease-relevant application, we studied the role of C4, recently implicated in schizophrenia, in engulfment of synaptic structures by human microglia. The ability to create complete patient-specific cellular models of critical microglial functions utilizing samples taken during a single clinical visit will extend the ability to model central nervous system disease while facilitating high-throughput screening.
Ann Neurol. 2016 Nov;80(5):730-740. doi: 10.1002/ana.24780. Epub 2016 Oct 19.
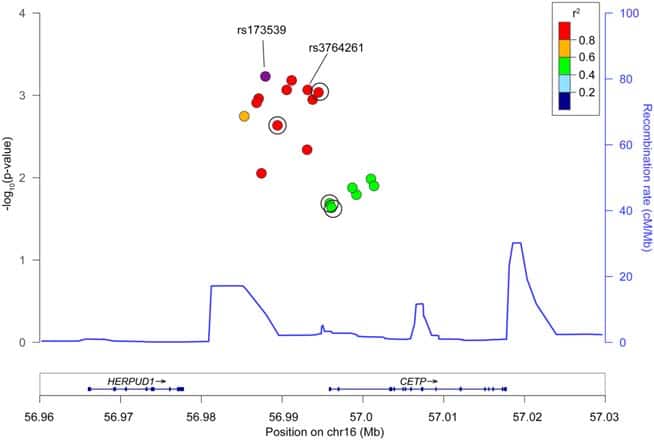
In observational epidemiologic studies, higher plasma high-density lipoprotein cholesterol (HDL-C) has been associated with increased risk of intracerebral hemorrhage (ICH). DNA sequence variants that decrease cholesteryl ester transfer protein (CETP) gene activity increase plasma HDL-C; as such, medicines that inhibit CETP and raise HDL-C are in clinical development. Here, we test the hypothesis that CETP DNA sequence variants associated with higher HDL-C also increase risk for ICH. Methods: We performed 2 candidate-gene analyses of CETP. First, we tested individual CETP variants in a discovery cohort of 1,149 ICH cases and 1,238 controls from 3 studies, followed by replication in 1,625 cases and 1,845 controls from 5 studies. Second, we constructed a genetic risk score comprised of 7 independent variants at the CETP locus and tested this score for association with HDL-C as well as ICH risk. Results: Twelve variants within CETP demonstrated nominal association with ICH, with the strongest association at the rs173539 locus (odds ratio [OR]51.25, standard error [SE]50.06, p56.031024) with no heterogeneity across studies (I250%). This association was replicated in patients of European ancestry (p50.03). A genetic score of CETP variants found to increase HDL-C by _2.85mg/dl in the Global Lipids Genetics Consortium was strongly associated with ICH risk (OR51.86, SE50.13, p51.3931026). Interpretation: Genetic variants in CETP associated with increased HDL-C raise the risk of ICH. Given ongoing therapeutic development in CETP inhibition and other HDL-raising strategies, further exploration of potential adverse cerebrovascular outcomes may be warranted.