Mol Psychiatry. 2016 Dec;21(12):1680-1689. doi: 10.1038/mp.2016.164. Epub 2016 Oct 11.
Schizophrenia is a devastating neurodevelopmental disorder with a complex genetic etiology. Widespread cortical gray matter loss has been observed in patients and prodromal samples. However, it remains unresolved whether schizophrenia-associated cortical structure variations arise due to disease etiology or secondary to the illness. Here we address this question using a partitioning-based heritability analysis of genome-wide single-nucleotide polymorphism (SNP) and neuroimaging data from 1750 healthy individuals. We find that schizophrenia-associated genetic variants explain a significantly enriched proportion of trait heritability in eight brain phenotypes (false discovery rate=10%). In particular, intracranial volume and left superior frontal gyrus thickness exhibit significant and robust associations with schizophrenia genetic risk under varying SNP selection conditions. Cross-disorder comparison suggests that the neurogenetic architecture of schizophrenia-associated brain regions is, at least in part, shared with other psychiatric disorders. Our study highlights key neuroanatomical correlates of schizophrenia genetic risk in the general population. These may provide fundamental insights into the complex pathophysiology of the illness, and a potential link to neurocognitive deficits shaping the disorder.
Richa Saxena, PhD Genome-wide association analyses of sleep disturbance traits identify new loci and highlight shared genetics with neuropsychiatric and metabolic traits
Chronic sleep disturbances, associated with cardiometabolic diseases, psychiatric disorders and all-cause mortality, affect 25-30% of adults worldwide. Although environmental factors contribute substantially to self-reported habitual sleep duration and disruption, these traits are heritable and identification of the genes involved should improve understanding of sleep, mechanisms linking sleep to disease and development of new therapies. We report single- and multiple-trait genome-wide association analyses of self-reported sleep duration, insomnia symptoms and excessive daytime sleepiness in the UK Biobank (n = 112,586). We discover loci associated with insomnia symptoms (near MEIS1, TMEM132E, CYCL1 and TGFBI in females and WDR27 in males), excessive daytime sleepiness (near AR-OPHN1) and a composite sleep trait (near PATJ (INADL) and HCRTR2) and replicate a locus associated with sleep duration (at PAX8). We also observe genetic correlation between longer sleep duration and schizophrenia risk (rg = 0.29, P = 1.90 × 10-13) and between increased levels of excessive daytime sleepiness and increased measures for adiposity traits (body mass index (BMI): rg = 0.20, P = 3.12 × 10-9; waist circumference: rg = 0.20, P = 2.12 × 10-7).
Ann Neurol. 2016 Nov;80(5):730-740. doi: 10.1002/ana.24780. Epub 2016 Oct 19.
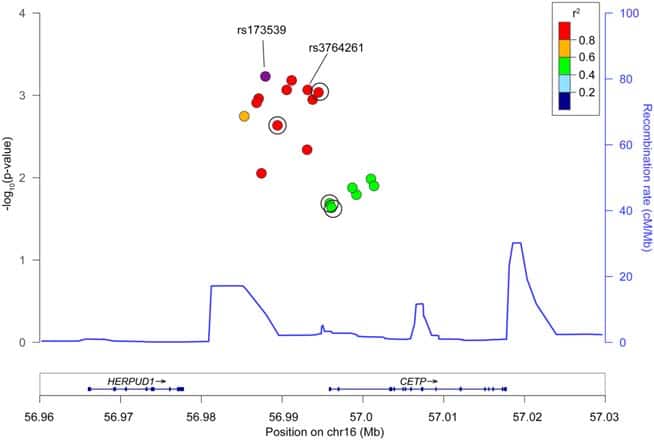
In observational epidemiologic studies, higher plasma high-density lipoprotein cholesterol (HDL-C) has been associated with increased risk of intracerebral hemorrhage (ICH). DNA sequence variants that decrease cholesteryl ester transfer protein (CETP) gene activity increase plasma HDL-C; as such, medicines that inhibit CETP and raise HDL-C are in clinical development. Here, we test the hypothesis that CETP DNA sequence variants associated with higher HDL-C also increase risk for ICH. Methods: We performed 2 candidate-gene analyses of CETP. First, we tested individual CETP variants in a discovery cohort of 1,149 ICH cases and 1,238 controls from 3 studies, followed by replication in 1,625 cases and 1,845 controls from 5 studies. Second, we constructed a genetic risk score comprised of 7 independent variants at the CETP locus and tested this score for association with HDL-C as well as ICH risk. Results: Twelve variants within CETP demonstrated nominal association with ICH, with the strongest association at the rs173539 locus (odds ratio [OR]51.25, standard error [SE]50.06, p56.031024) with no heterogeneity across studies (I250%). This association was replicated in patients of European ancestry (p50.03). A genetic score of CETP variants found to increase HDL-C by _2.85mg/dl in the Global Lipids Genetics Consortium was strongly associated with ICH risk (OR51.86, SE50.13, p51.3931026). Interpretation: Genetic variants in CETP associated with increased HDL-C raise the risk of ICH. Given ongoing therapeutic development in CETP inhibition and other HDL-raising strategies, further exploration of potential adverse cerebrovascular outcomes may be warranted.
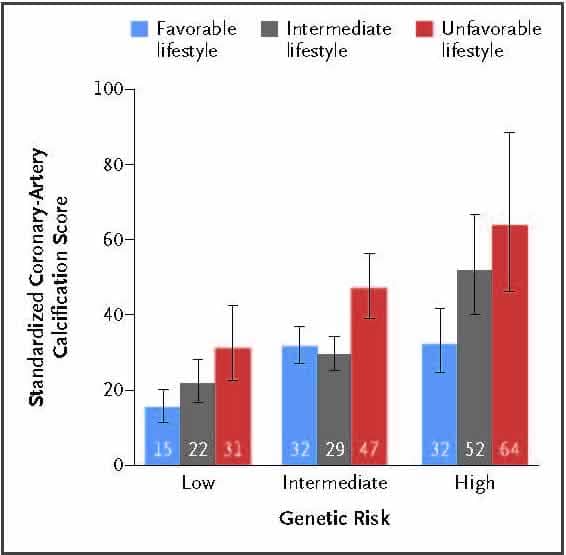
Sekar Kathiresan, MD Genetic Risk, Adherence to a Healthy Lifestyle, and Coronary Disease
N Engl J Med November 13, 2016. DOI: 10.1056/NEJMoa1605086
Both genetic and lifestyle factors contribute to individual-level risk of coronary artery disease. The extent to which increased genetic risk can be offset by a healthy lifestyle is unknown. Across four studies involving 55,685 participants, genetic and lifestyle factors were independently associated with susceptibility to coronary artery disease. Among par- ticipants at high genetic risk, a favorable lifestyle was associated with a nearly 50% lower relative risk of coronary artery disease than was an unfavorable lifestyle. (Funded by the National Institutes of Health and others.)
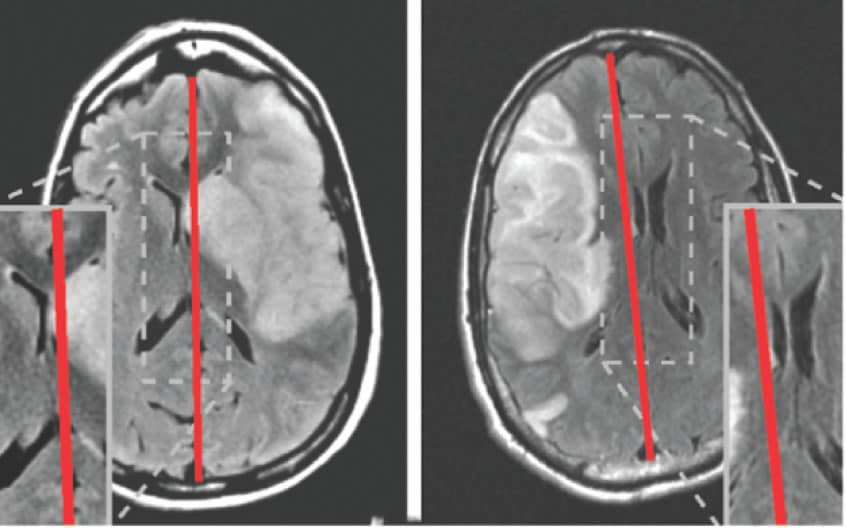
W. Taylor Kimberly, MD, PhD Safety and efficacy of intravenous glyburide on brain swelling after large hemispheric infarction (GAMES-RP): a randomised, double-blind, placebo-controlled phase 2 trial
Lancet Neurol. 2016 Oct;15(11):1160-9. doi: 10.1016/S1474-4422(16)30196-X. Epub 2016 Aug 23.
Preclinical models of stroke have shown that intravenous glyburide reduces brain swelling and improves survival. We assessed whether intravenous glyburide (RP-1127; glibenclamide) would safely reduce brain swelling, decrease the need for decompressive craniectomy, and improve clinical outcomes in patients presenting with a large hemispheric infarction. Intravenous glyburide was well tolerated in patients with large hemispheric stroke at risk for cerebral oedema. There was no difference in the composite primary outcome. Further study is warranted to assess the potential clinical benefit of a reduction in swelling by intravenous glyburide.
An accurate accounting of persons with Down syndrome (DS) has remained elusive because no population-based registries exist in the United States. The purpose of this study was to estimate this population size by age, race, and ethnicity. Until 2008, DS was a rare disease. In more recent decades, the population growth of people with DS has leveled off for non-Hispanic whites as a consequence of elective terminations. Changes in childhood survival have impacted the age distribution of people with DS, with more people in their fourth, fifth, and sixth decades of life.
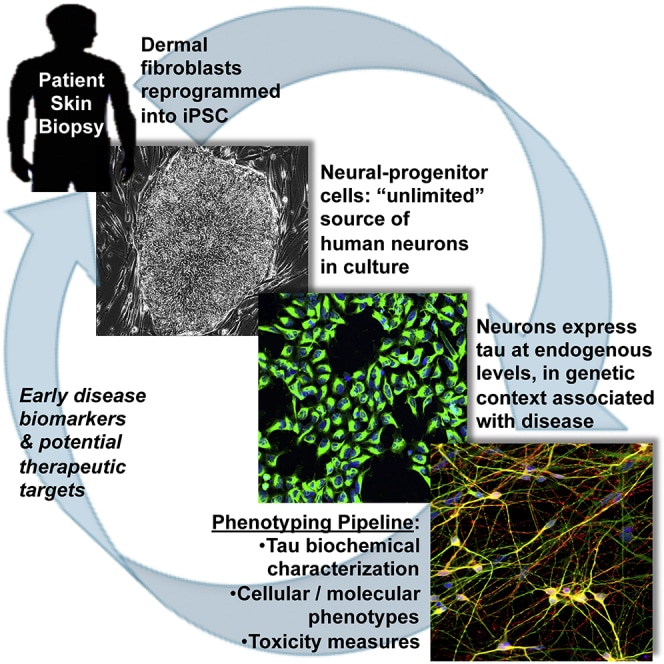
Stephen J. Haggarty, PhD Human iPSC-Derived Neuronal Model of Tau-A152T Frontotemporal Dementia Reveals Tau-Mediated Mechanisms of Neuronal Vulnerability
Frontotemporal dementia (FTD) and other tauopathies characterized by focal brain neurodegeneration and pathological accumulation of proteins are commonly associated with tau mutations. However, the mechanismof neuronal loss is not fully understood. To identify molecular events associated with tauopathy, we studied induced pluripotent stem cell (iPSC)-derived neurons from individuals carrying the tau-A152T variant.We highlight the potential of in-depth phenotyping of human neuronal cell models for pre-clinical studies and identification of modulators of endogenous tau toxicity. Through a panel of biochemical and cellular assays, A152T neurons showed accumulation, redistribution, and decreased solubility of tau. Upregulation of tau was coupled to enhanced stress-inducible markers and cell vulnerability to proteotoxic, excitotoxic, and mitochondrial stressors, which was rescued upon CRISPR/Cas9-mediated targeting of tau or by pharmacological activation of autophagy. Our findings unmask tau-mediated perturbations of specific pathways associated with neuronal vulnerability, revealing potential early disease biomarkers and therapeutic targets for FTD and other tauopathies.
Nature. 2016 Aug 18;536(7616):285-91. doi: 10.1038/nature19057.
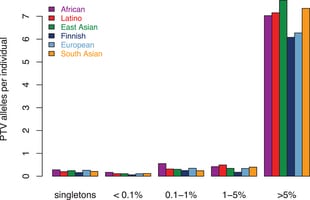
Large-scale reference data sets of human genetic variation are critical for the medical and functional interpretation of DNA sequence changes. Here we describe the aggregation and analysis of high-quality exome (protein-coding region) DNA sequence data for 60,706 individuals of diverse ancestries generated as part of the Exome Aggregation Consortium (ExAC). This catalogue of human genetic diversity contains an average of one variant every eight bases of the exome, and provides direct evidence for the presence of widespread mutational recurrence. We have used this catalogue to calculate objective metrics of pathogenicity for sequence variants, and to identify genes subject to strong selection against various classes of mutation; identifying 3,230 genes with near-complete depletion of predicted protein-truncating variants, with 72% of these genes having no currently established human disease phenotype. Finally, we demonstrate that these data can be used for the efficient filtering of candidate disease-causing variants, and for the discovery of human ‘knockout’ variants in protein-coding genes.
Nat Genet. 2016 Aug;48(8):856-66. doi: 10.1038/ng.3598. Epub 2016 Jun 20.
Migraine is a debilitating neurological disorder affecting around one in seven people worldwide, but its molecular mechanisms remain poorly understood. There is some debate about whether migraine is a disease of vascular dysfunction or a result of neuronal dysfunction with secondary vascular changes. Genome-wide association (GWA) studies have thus far identified 13 independent loci associated with migraine. To identify new susceptibility loci, we carried out a genetic study of migraine on 59,674 affected subjects and 316,078 controls from 22 GWA studies. We identified 44 independent single-nucleotide polymorphisms (SNPs) significantly associated with migraine risk (P < 5 × 10(-8)) that mapped to 38 distinct genomic loci, including 28 loci not previously reported and a locus that to our knowledge is the first to be identified on chromosome X. In subsequent computational analyses, the identified loci showed enrichment for genes expressed in vascular and smooth muscle tissues, consistent with a predominant theory of migraine that highlights vascular etiologies.
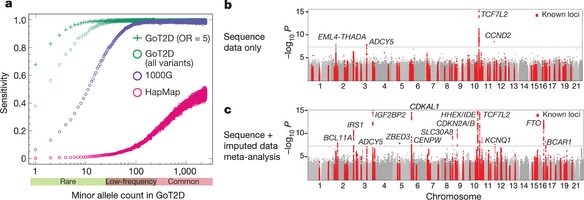
The genetic architecture of common traits, including the number, frequency, and effect sizes of inherited variants that contribute to individual risk, has been long debated. Genome-wide association studies have identified scores of common variants associated with type 2 diabetes, but in aggregate, these explain only a fraction of the heritability of this disease. Here, to test the hypothesis that lower-frequency variants explain much of the remainder, the GoT2D and T2D-GENES consortia performed whole-genome sequencing in 2,657 European individuals with and without diabetes, and exome sequencing in 12,940 individuals from five ancestry groups. To increase statistical power, we expanded the sample size via genotyping and imputation in a further 111,548 subjects. Variants associated with type 2 diabetes after sequencing were overwhelmingly common and most fell within regions previously identified by genome-wide association studies. Comprehensive enumeration of sequence variation is necessary to identify functional alleles that provide important clues to disease pathophysiology, but large-scale sequencing does not support the idea that lower-frequency variants have amajor role in predisposition to type 2 diabetes.