Genome-wide association studies (GWAS) have made little progress in identifying variants linked to depression. We hypothesized that examining depressive symptoms and considering gene-environment interaction (GxE) might improve efficiency for gene discovery. We therefore conducted a GWAS and genome-wide by environment interaction study (GWEIS) of depressive symptoms. Our results underscore the need for larger samples, more GEWIS, and greater investigation into genetic and environmental determinants of depressive symptoms in minorities.
Science. 2016 Apr 1;352(6281):54-61. doi: 10.1126/science.aad9642. Epub 2016 Feb 25.
Defects in the mitochondrial respiratory chain (RC) underlie a spectrum of human conditions, ranging from devastating inborn errors of metabolism to aging. We performed a genome-wide Cas9-mediated screen to identify factors that are protective during RC inhibition. Our results highlight the hypoxia response, an endogenous program evolved to adapt to limited oxygen availability. Genetic or small-molecule activation of the hypoxia response is protective against mitochondrial toxicity in cultured cells and zebrafish models. Chronic hypoxia leads to a marked improvement in survival, body weight, body temperature, behavior, neuropathology, and disease biomarkers in a genetic mouse model of Leigh syndrome, the most common pediatric manifestation of mitochondrial disease. Further preclinical studies are required to assess whether hypoxic exposure can be developed into a safe and effective treatment for human diseases associated with mitochondrial dysfunction.
Evidence that different neuropsychiatric conditions share genetic liability has increased interest in phenotypes with ‘cross-disorder’ relevance, as they may contribute to revised models of psychopathology. Cognition is a promising construct for study; yet, evidence that the same cognitive functions are impaired across different forms of psychopathology comes primarily from separate studies of individual categorical diagnoses versus controls. Given growing support for dimensional models that cut across traditional diagnostic boundaries, we aimed to determine, within a single cohort, whether performance on measures of executive functions (EFs) predicted dimensions of different psychopathological conditions known to share genetic liability. Findings extend support for the relevance of cognition to neuropsychiatric conditions that share underlying genetic risk. They suggest that higher-order cognition, including EFs, relates to the dimensional spectrum of each of these disorders and not just the clinical diagnoses. Moreover, results have implications for bottom-up models linking genes, cognition, and a general psychopathology liability.
Ihn Sik Seong, PhD, Jong-Min Lee, PhD Huntingtin’s spherical solenoid structure enables polyglutamine tract-dependent modulation of its structure and function
Elife. 2016 Mar 22;5:e11184. doi: 10.7554/eLife.11184
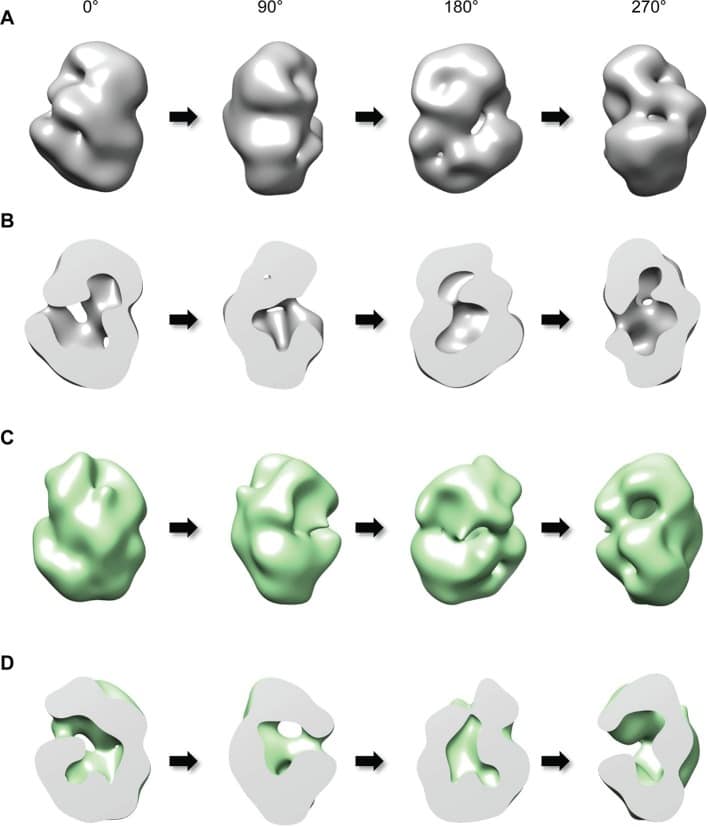
The polyglutamine expansion in huntingtin protein causes Huntington’s disease. Here, we investigated structural and biochemical properties of huntingtin and the effect of the polyglutamine expansion using various biophysical experiments including circular dichroism, single-particle electron microscopy and cross-linking mass spectrometry. Huntingtin is likely composed of five distinct domains and adopts a spherical α-helical solenoid where the amino-terminal and carboxyl-terminal regions fold to contain a circumscribed central cavity. Interestingly, we showed that the polyglutamine expansion increases α-helical properties of huntingtin and affects the intramolecular interactions among the domains. Our work delineates the structural characteristics of full-length huntingtin, which are affected by the polyglutamine expansion, and provides an elegant solution to the apparent conundrum of how the extreme amino-terminal polyglutamine tract confers a novel property on huntingtin, causing the disease.
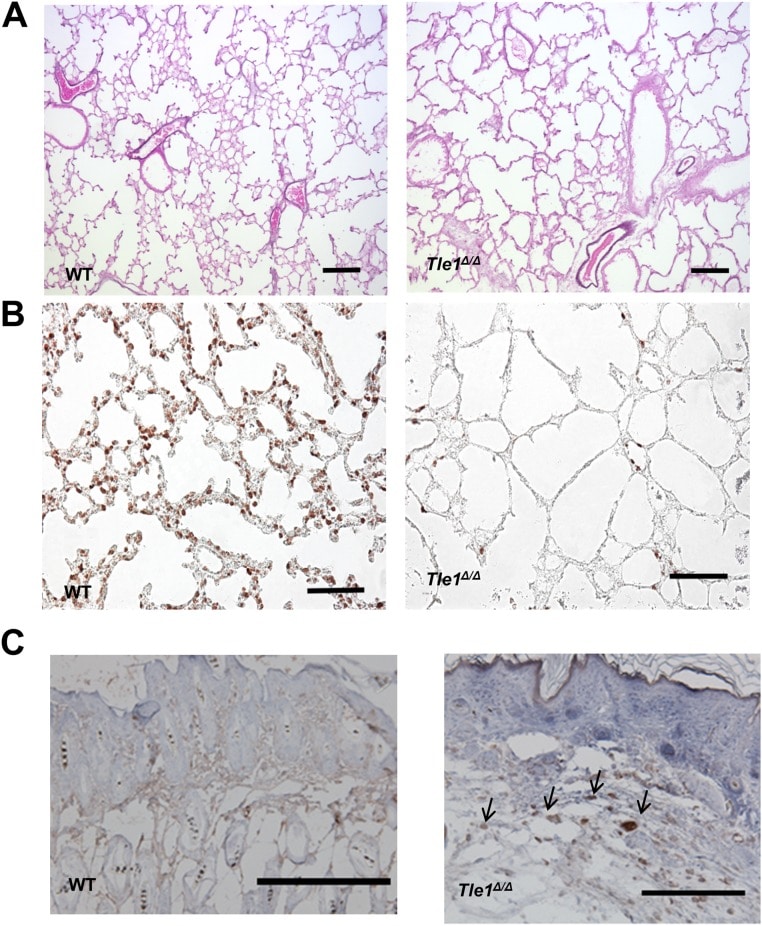
Tle1 (transducin-like enhancer of split 1) is a corepressor that interacts with a variety of DNA-binding transcription factors and has been implicated in many cellular functions; however, physiological studies are limited. Tle1-deficient (Tle1(Δ/Δ)) mice, although grossly normal at birth, exhibit skin defects, lung hypoplasia, severe runting, poor body condition, and early mortality. Tle1(Δ/Δ) mice display a chronic inflammatory phenotype with increased expression of inflammatory cytokines and chemokines in the skin, lung, and intestine and increased circulatory IL-6 and G-CSF, along with a hematopoietic shift toward granulocyte macrophage progenitor and myeloid cells. Tle1(Δ/Δ) macrophages produce increased inflammatory cytokines in response to Toll-like receptor (TLR) agonists and lipopolysaccharides (LPS), and Tle1(Δ/Δ) mice display an enhanced inflammatory response to ear skin 12-O-tetradecanoylphorbol-13-acetate treatment. Loss of Tle1 not only results in increased phosphorylation and activation of proinflammatory NF-κB but also results in decreased Hes1 (hairy and enhancer of split-1), a negative regulator of inflammation in macrophages. Furthermore, Tle1(Δ/Δ) mice exhibit accelerated growth of B6-F10 melanoma xenografts. Our work provides the first in vivo evidence, to our knowledge, that TLE1 is a major counterregulator of inflammation with potential roles in a variety of inflammatory diseases and in cancer progression.
Jonathan Rosand, MD, MSc Loci associated with ischaemic stroke and its subtypes (SiGN): a genome-wide association study
Lancet Neurol. 2015 Dec 18. pii: S1474-4422(15)00338-5. doi: 10.1016/S1474-4422(15)00338-5.
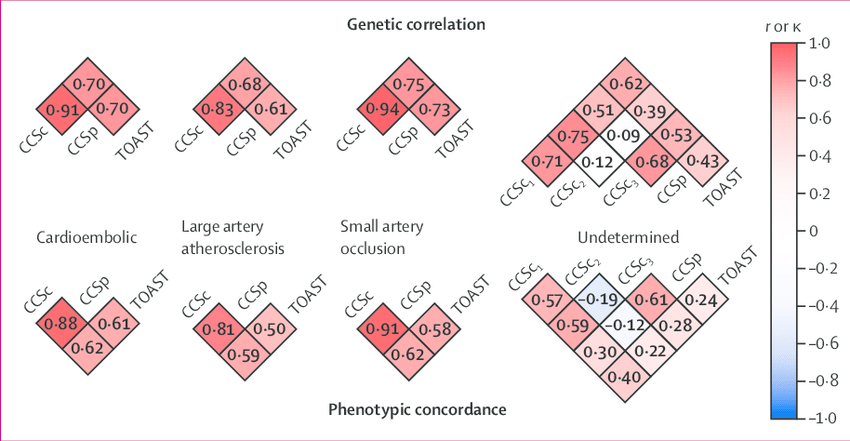
The discovery of disease-associated loci through genome-wide association studies (GWAS) is the leading genetic approach to the identification of novel biological pathways underlying diseases in humans. Until recently, GWAS in ischaemic stroke have been limited by small sample sizes and have yielded few loci associated with ischaemic stroke. We did a large-scale GWAS to identify additional susceptibility genes for stroke and its subtypes. Our results suggest that all ischaemic stroke-related loci previously implicated by GWAS are subtype specific. We identified a novel gene associated with large artery atherosclerosis stroke susceptibility. Follow-up studies will be necessary to establish whether the locus near TSPAN2 can be a target for a novel therapeutic approach to stroke prevention. In view of the subtype-specificity of the associations detected, the rich phenotyping data available in the Stroke Genetics Network (SiGN) are likely to be crucial for further genetic discoveries related to ischaemic stroke.
X-linked adrenoleukodystrophy is caused by mutations in the ABCD1 gene leading to accumulation of very long chain fatty acids. Its most severe neurological manifestation is cerebral adrenoleukodystrophy. Here we demonstrate that progressive inflammatory demyelination in cerebral adrenoleukodystrophy coincides with blood–brain barrier dysfunction, increased MMP9 expression, and changes in endothelial tight junction proteins as well as adhesion molecules. ABCD1, but not its closest homologue ABCD2, is highly expressed in human brain microvascular endothelial cells, far exceeding its expression in the systemic vasculature. Silencing of ABCD1 in human brain microvascular endothelial cells causes accumulation of very long chain fatty acids, but much later than the immediate upregulation of adhesion molecules and decrease in tight junction proteins. This results in greater adhesion and transmigration of monocytes across the endothelium. PCR-array screening of human brain microvascular endothelial cells after ABCD1 silencing revealed downregulation of both mRNA and protein levels of the transcription factor c-MYC (encoded by MYC). Interestingly, MYC silencing mimicked the effects of ABCD1 silencing on CLDN5 and ICAM1 without decreasing the levels of ABCD1 protein itself. Together, these data demonstrate that ABCD1 deficiency induces significant alterations in brain endothelium via c-MYC and may thereby contribute to the increased trafficking of leucocytes across the blood–brain barrier as seen in cerebral adrenouleukodystrophy.
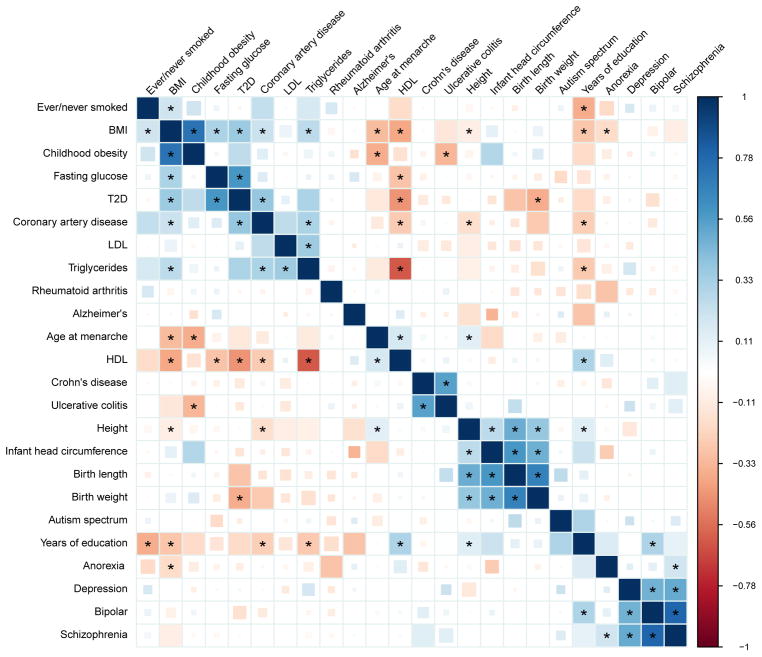
Identifying genetic correlations between complex traits and diseases can provide useful etiological insights and help prioritize likely causal relationships. The major challenges preventing estimation of genetic correlation from genome-wide association study (GWAS) data with current methods are the lack of availability of individual-level genotype data and widespread sample overlap among meta-analyses. We circumvent these difficulties by introducing a technique—cross-trait LD Score regression—for estimating genetic correlation that requires only GWAS summary statistics and is not biased by sample overlap. We use this method to estimate 276 genetic correlations among 24 traits. The results include genetic correlations between anorexia nervosa and schizophrenia, anorexia and obesity, and educational attainment and several diseases. These results highlight the power of genome-wide analyses, as there currently are no significantly associated SNPs for anorexia nervosa and only three for educational attainment
Nature. 2015 Sep 3;525(7567):109-13. doi: 10.1038/nature14670. Epub 2015 Aug 10.
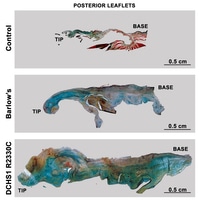
Mitral valve prolapse (MVP) is a common cardiac valve disease that affects nearly 1 in 40 individuals1–3. It can manifest as mitral regurgitation and is the leading indication for mitral valve surgery4,5. Despite a clear heritable component, the genetic aetiology leading to non-syndromic MVP has remained elusive. Four affected individuals from a large multigenerational family segregating non-syndromic MVP underwent capture sequencing of the linked interval on chromosome 11. We report a missense mutation in the DCHS1 gene, the human homologue of the Drosophila cell polarity gene dachsous (ds), that segregates with MVP in the family. Morpholino knockdown of the zebrafish homologue dachsous1b resulted in a cardiac atrioventricular canal defect that could be rescued by wild-type humanDCHS1, but not byDCHS1 messenger RNA with the familial mutation. Further genetic studies identified two additional families in which a second deleterious DCHS1 mutation segregates with MVP. Both DCHS1 mutations reduce protein stability as demonstrated in zebrafish, cultured cells and, notably, in mitral valve interstitial cells (MVICs) obtained during mitral valve repair surgery of a proband. Dchs11/2 mice had prolapse of thickened mitral leaflets, which could be traced back to developmental errors in valve morphogenesis. DCHS1 deficiency in MVP patient MVICs, as well as in Dchs11/2 mouse MVICs, result in altered migration and cellular patterning, supporting these processes as aetiological underpinnings for the disease. Understanding the role ofDCHS1 in mitral valve development and MVP pathogenesis holds potential for therapeutic insights for this very common disease.
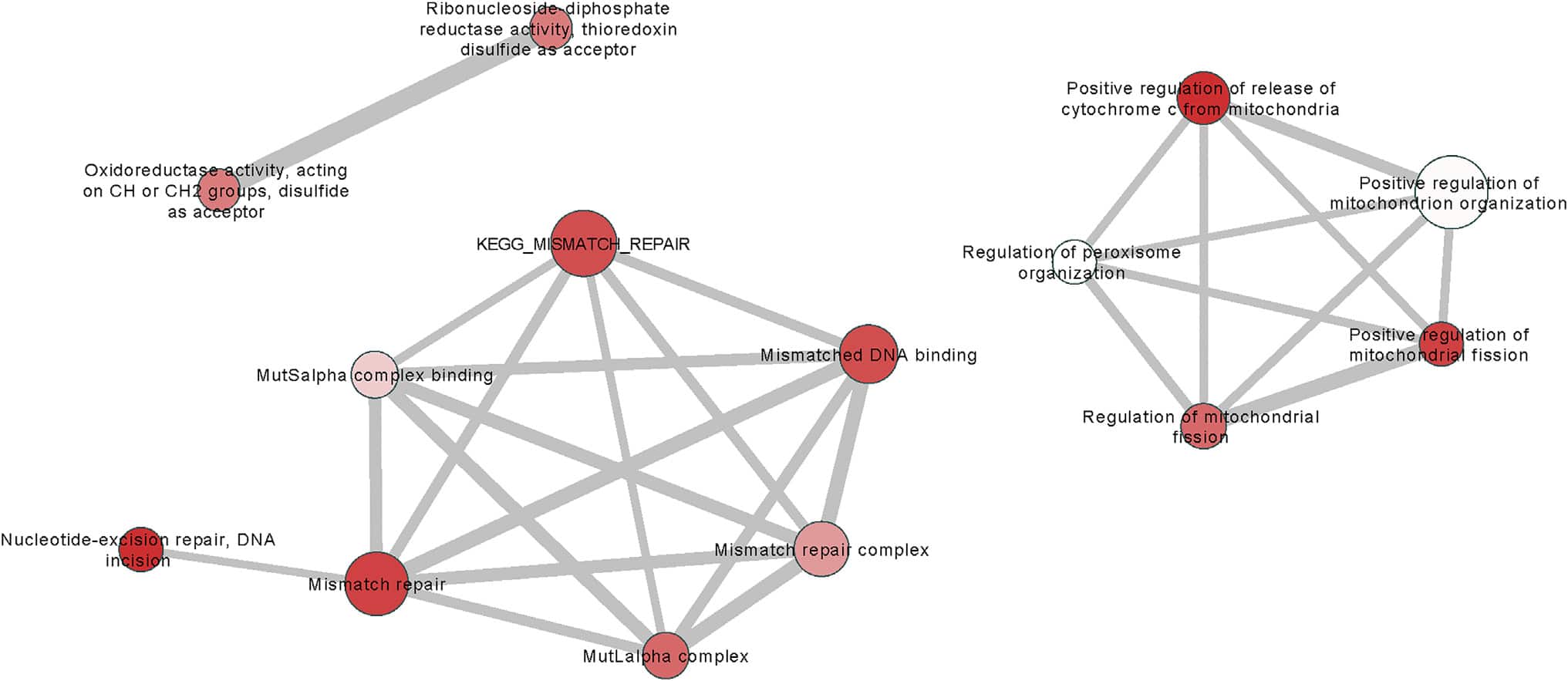
As a Mendelian neurodegenerative disorder, the genetic risk of Huntington’s disease (HD) is conferred entirely by an HTT CAG repeat expansion whose length is the primary determinant of the rate of pathogenesis leading to disease onset. To investigate the pathogenic process that precedes disease, we used genome-wide association (GWA) analysis to identify loci harboring genetic variations that alter the age at neurological onset of HD. A chromosome 15 locus displays two independent effects that accelerate or delay onset by 6.1 years and 1.4 years, respectively, whereas a chromosome 8 locus hastens onset by 1.6 years. Association at MLH1 and pathway analysis of the full GWA results support a role for DNA handling and repair mechanisms in altering the course of HD. Our findings demonstrate that HD disease modification in humans occurs in nature and offer a genetic route to identifying in-human validated therapeutic targets in this and other Mendelian disorders.