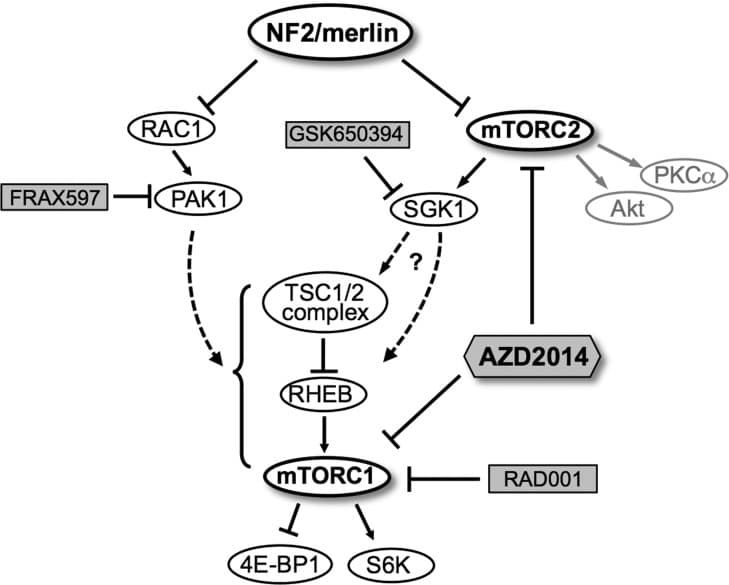
Meningiomas are the most common primary intracranial adult tumor. All Neurofibromatosis 2 (NF2)-associated meningiomas and ~60% of sporadic meningiomas show loss of NF2 tumor suppressor protein. There are no effective medical therapies for progressive and recurrent meningiomas. Our previous work demonstrated aberrant activation of mTORC1 signaling that led to ongoing clinical trials with rapamycin analogs for NF2 and sporadic meningioma patients. Here we performed a high-throughput kinome screen to identify kinases responsible for mTORC1 pathway activation in NF2-deficient meningioma cells. Among the emerging top candidates were the mTORC2-specific target serum/glucocorticoid-regulated kinase 1 (SGK1) and p21-activated kinase 1 (PAK1). In NF2-deficient meningioma cells, inhibition of SGK1 rescues mTORC1 activation, and SGK1 activation is sensitive to dual mTORC1/2 inhibitor AZD2014, but not to rapamycin. PAK1 inhibition also leads to attenuated mTORC1 but not mTORC2 signaling, suggesting that mTORC2/SGK1 and Rac1/PAK1 pathways are independently responsible for mTORC1 activation in NF2-deficient meningiomas. Using CRISPR-Cas9 genome editing, we generated isogenic human arachnoidal cell lines (ACs), the origin cell type for meningiomas, expressing or lacking NF2. NF2-null CRISPR ACs recapitulate the signaling of NF2-deficient meningioma cells. Interestingly, we observe increased SGK1 transcription and protein expression in NF2-CRISPR ACs and in primary NF2-negative meningioma lines. Moreover, we demonstrate that the dual mTORC1/mTORC2 inhibitor, AZD2014 is superior to rapamycin and PAK inhibitor FRAX597 in blocking proliferation of meningioma cells. Importantly, AZD2014 is currently in use in several clinical trials of cancer. Therefore, we believe that AZD2014 may provide therapeutic advantage over rapalogs for recurrent and progressive meningiomas.
Susan L. Cotman, PhD, Stephen J. Haggarty, PhD Unbiased Cell-based Screening in a Neuronal Cell Model of Batten Disease Highlights an Interaction between Ca2+ Homeostasis, Autophagy, and CLN3 Protein Function
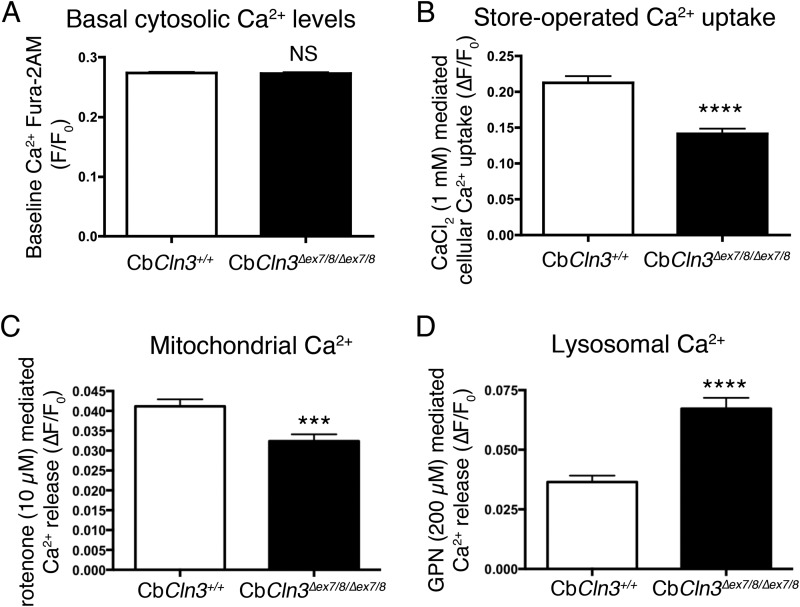
Abnormal accumulation of undigested macromolecules, often disease-specific, is a major feature of lysosomal and neurodegenerative disease and is frequently attributed to defective autophagy. The mechanistic underpinnings of the autophagy defects are the subject of intense research, which is aided by genetic disease models. To gain an improved understanding of the pathways regulating defective autophagy specifically in juvenile neuronal ceroid lipofuscinosis (JNCL or Batten disease), a neurodegenerative disease of childhood, we developed and piloted a GFP-microtubule-associated protein 1 light chain 3 (GFP-LC3) screening assay to identify, in an unbiased fashion, genotype-sensitive small molecule autophagy modifiers, employing a JNCL neuronal cell model bearing the most common disease mutation in CLN3. Thapsigargin, a sarco/endoplasmic reticulum Ca(2+)-ATPase (SERCA) Ca(2+) pump inhibitor, reproducibly displayed significantly more activity in the mouse JNCL cells, an effect that was also observed in human-induced pluripotent stem cell-derived JNCL neural progenitor cells. The mechanism of thapsigargin sensitivity was Ca(2+)-mediated, and autophagosome accumulation in JNCL cells could be reversed by Ca(2+) chelation. Interrogation of intracellular Ca(2+) handling highlighted alterations in endoplasmic reticulum, mitochondrial, and lysosomal Ca(2+) pools and in store-operated Ca(2+) uptake in JNCL cells. These results further support an important role for the CLN3 protein in intracellular Ca(2+) handling and in autophagic pathway flux and establish a powerful new platform for therapeutic screening.
ACS Chem Biol. 2015 Mar 20;10(3):883-90. doi: 10.1021/cb500838r. Epub 2015 Jan 8
We examined the effects of isoform-specific histone deacetylase (HDAC) inhibitors on β-catenin posttranslational modifications in neural progenitor cells (NPCs) derived from human induced pluripotent stem cells (iPSCs). β-catenin is a multifunctional protein with important roles in the developing and adult central nervous system. Activation of the Wnt pathway results in stabilization and nuclear translocation of β-catenin, resulting in activation of multiple target genes. In addition, β-catenin forms a complex with cadherins at the plasma membrane as part of the adherens junctions. The N-terminus of β-catenin has phosphorylation, ubiquitination, and acetylation sites that regulate its stability and signaling. In the absence of a Wnt signal, Ser33, Ser37, and Thr41 are constitutively phosphorylated by glycogen synthase kinase 3β (GSK3β). β-Catenin phosphorylated at these sites is recognized by β-transducin repeat-containing protein (βTrCP), which results in ubiquitination and degradation by the ubiquitin-proteasome pathway. The N-terminal regulatory domain of β-catenin also includes Ser45, a phosphorylation site for Casein Kinase 1α (CK1α) and Lys49, which is acetylated by the acetyltransferase p300/CBP-associated factor (PCAF). The relevance of Lys49 acetylation and Ser45 phosphorylation to the function of β-catenin is an active area of investigation. We find that HDAC6 inhibitors increase Lys49 acetylation and Ser45 phosphorylation but do not affect Ser33, Ser37, and Thr41 phosphorylation. Lys49 acetylation results in decreased ubiquitination of β-catenin in the presence of proteasome inhibition. While increased Lys49 acetylation does not affect total levels of β-catenin, it results in increased membrane localization of β-catenin.
Nat Genet. 2014 Aug;46(8):826-36. doi: 10.1038/ng.3014. Epub 2014 Jun 22.
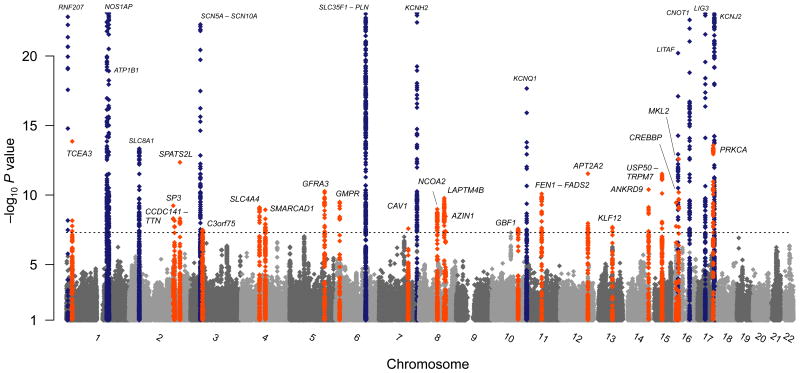
The QT interval, an electrocardiographic measure reflecting myocardial repolarization, is a heritable trait. QT prolongation is a risk factor for ventricular arrhythmias and sudden cardiac death (SCD) and could indicate the presence of the potentially lethal mendelian long-QT syndrome (LQTS). Using a genome-wide association and replication study in up to 100,000 individuals, we identified 35 common variant loci associated with QT interval that collectively explain ~8–10% of QT-interval variation and highlight the importance of calcium regulation in myocardial repolarization. Rare variant analysis of 6 new QT interval–associated loci in 298 unrelated probands with LQTS identified coding variants not found in controls but of uncertain causality and therefore requiring validation. Several newly identified loci encode proteins that physically interact with other recognized repolarization proteins. Our integration of common variant association, expression and orthogonal protein-protein interaction screens provides new insights into cardiac electrophysiology and identifies new candidate genes for ventricular arrhythmias, LQTS and SCD.
James Walker, PhD Genetic and Functional Studies Implicate Synaptic Overgrowth and Ring Gland cAMP/PKA Signaling Defects in the Drosophila melanogaster Neurofibromatosis-1 Growth Deficiency
PLoS Genet. 2013 Nov;9(11):e1003958. doi: 10.1371/journal.pgen.1003958. Epub 2013 Nov 21.
Neurofibromatosis type 1 (NF1), a genetic disease that affects 1 in 3,000, is caused by loss of a large evolutionary conserved protein that serves as a GTPase Activating Protein (GAP) for Ras. Among Drosophila melanogaster Nf1 (dNf1) null mutant phenotypes, learning/memory deficits and reduced overall growth resemble human NF1 symptoms. These and other dNf1 defects are relatively insensitive to manipulations that reduce Ras signaling strength but are suppressed by increasing signaling through the 3′-5′ cyclic adenosine monophosphate (cAMP) dependent Protein Kinase A (PKA) pathway, or phenocopied by inhibiting this pathway. However, whether dNf1 affects cAMP/PKA signaling directly or indirectly remains controversial. To shed light on this issue we screened 486 1(st) and 2(nd) chromosome deficiencies that uncover >80% of annotated genes for dominant modifiers of the dNf1 pupal size defect, identifying responsible genes in crosses with mutant alleles or by tissue-specific RNA interference (RNAi) knockdown. Validating the screen, identified suppressors include the previously implicated dAlk tyrosine kinase, its activating ligand jelly belly (jeb), two other genes involved in Ras/ERK signal transduction and several involved in cAMP/PKA signaling. Novel modifiers that implicate synaptic defects in the dNf1 growth deficiency include the intersectin-related synaptic scaffold protein Dap160 and the cholecystokinin receptor-related CCKLR-17D1 drosulfakinin receptor. Providing mechanistic clues, we show that dAlk, jeb and CCKLR-17D1 are among mutants that also suppress a recently identified dNf1 neuromuscular junction (NMJ) overgrowth phenotype and that manipulations that increase cAMP/PKA signaling in adipokinetic hormone (AKH)-producing cells at the base of the neuroendocrine ring gland restore the dNf1 growth deficiency. Finally, supporting our previous contention that ALK might be a therapeutic target in NF1, we report that human ALK is expressed in cells that give rise to NF1 tumors and that NF1 regulated ALK/RAS/ERK signaling appears conserved in man.
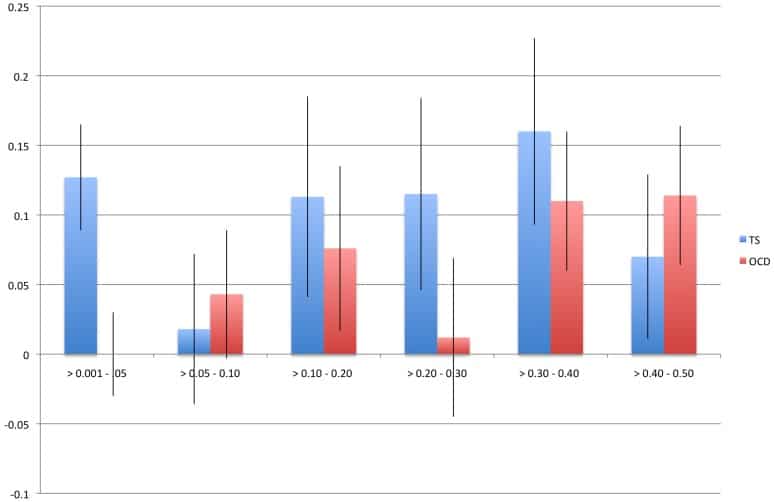
The direct estimation of heritability from genome-wide common variant data as implemented in the program Genome-wide Complex Trait Analysis (GCTA) has provided a means to quantify heritability attributable to all interrogated variants. We have quantified the variance in liability to disease explained by all SNPs for two phenotypically-related neurobehavioral disorders, obsessive-compulsive disorder (OCD) and Tourette Syndrome (TS), using GCTA. Our analysis yielded a heritability point estimate of 0.58 (se = 0.09, p = 5.64e-12) for TS, and 0.37 (se = 0.07, p = 1.5e-07) for OCD. In addition, we conducted multiple genomic partitioning analyses to identify genomic elements that concentrate this heritability. We examined genomic architectures of TS and OCD by chromosome, MAF bin, and functional annotations. In addition, we assessed heritability for early onset and adult onset OCD. Among other notable results, we found that SNPs with a minor allele frequency of less than 5% accounted for 21% of the TS heritability and 0% of the OCD heritability. Additionally, we identified a significant contribution to TS and OCD heritability by variants significantly associated with gene expression in two regions of the brain (parietal cortex and cerebellum) for which we had available expression quantitative trait loci (eQTLs). Finally we analyzed the genetic correlation between TS and OCD, revealing a genetic correlation of 0.41 (se = 0.15, p = 0.002). These results are very close to previous heritability estimates for TS and OCD based on twin and family studies, suggesting that very little, if any, heritability is truly missing (i.e., unassayed) from TS and OCD GWAS studies of common variation. The results also indicate that there is some genetic overlap between these two phenotypically-related neuropsychiatric disorders, but suggest that the two disorders have distinct genetic architectures.
PLoS Genet. 2013 Oct;9(10):e1003930. doi: 10.1371/journal.pgen.1003930. Epub 2013 Oct 31.
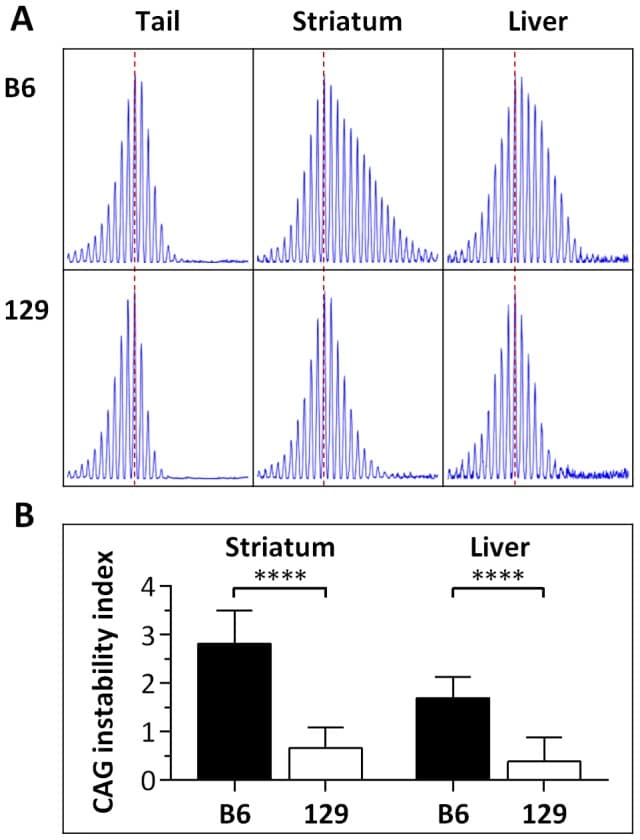
The Huntington’s disease gene (HTT) CAG repeat mutation undergoes somatic expansion that correlates with pathogenesis. Modifiers of somatic expansion may therefore provide routes for therapies targeting the underlying mutation, an approach that is likely applicable to other trinucleotide repeat diseases. Huntington’s disease Hdh(Q111) mice exhibit higher levels of somatic HTT CAG expansion on a C57BL/6 genetic background (B6.Hdh(Q111) ) than on a 129 background (129.Hdh(Q111) ). Linkage mapping in (B6x129).Hdh(Q111) F2 intercross animals identified a single quantitative trait locus underlying the strain-specific difference in expansion in the striatum, implicating mismatch repair (MMR) gene Mlh1 as the most likely candidate modifier. Crossing B6.Hdh(Q111) mice onto an Mlh1 null background demonstrated that Mlh1 is essential for somatic CAG expansions and that it is an enhancer of nuclear huntingtin accumulation in striatal neurons. Hdh(Q111) somatic expansion was also abolished in mice deficient in the Mlh3 gene, implicating MutLγ (MLH1-MLH3) complex as a key driver of somatic expansion. Strikingly, Mlh1 and Mlh3 genes encoding MMR effector proteins were as critical to somatic expansion as Msh2 and Msh3 genes encoding DNA mismatch recognition complex MutSβ (MSH2-MSH3). The Mlh1 locus is highly polymorphic between B6 and 129 strains. While we were unable to detect any difference in base-base mismatch or short slipped-repeat repair activity between B6 and 129 MLH1 variants, repair efficiency was MLH1 dose-dependent. MLH1 mRNA and protein levels were significantly decreased in 129 mice compared to B6 mice, consistent with a dose-sensitive MLH1-dependent DNA repair mechanism underlying the somatic expansion difference between these strains. Together, these data identify Mlh1 and Mlh3 as novel critical genetic modifiers of HTT CAG instability, point to Mlh1 genetic variation as the likely source of the instability difference in B6 and 129 strains and suggest that MLH1 protein levels play an important role in driving of the efficiency of somatic expansions.
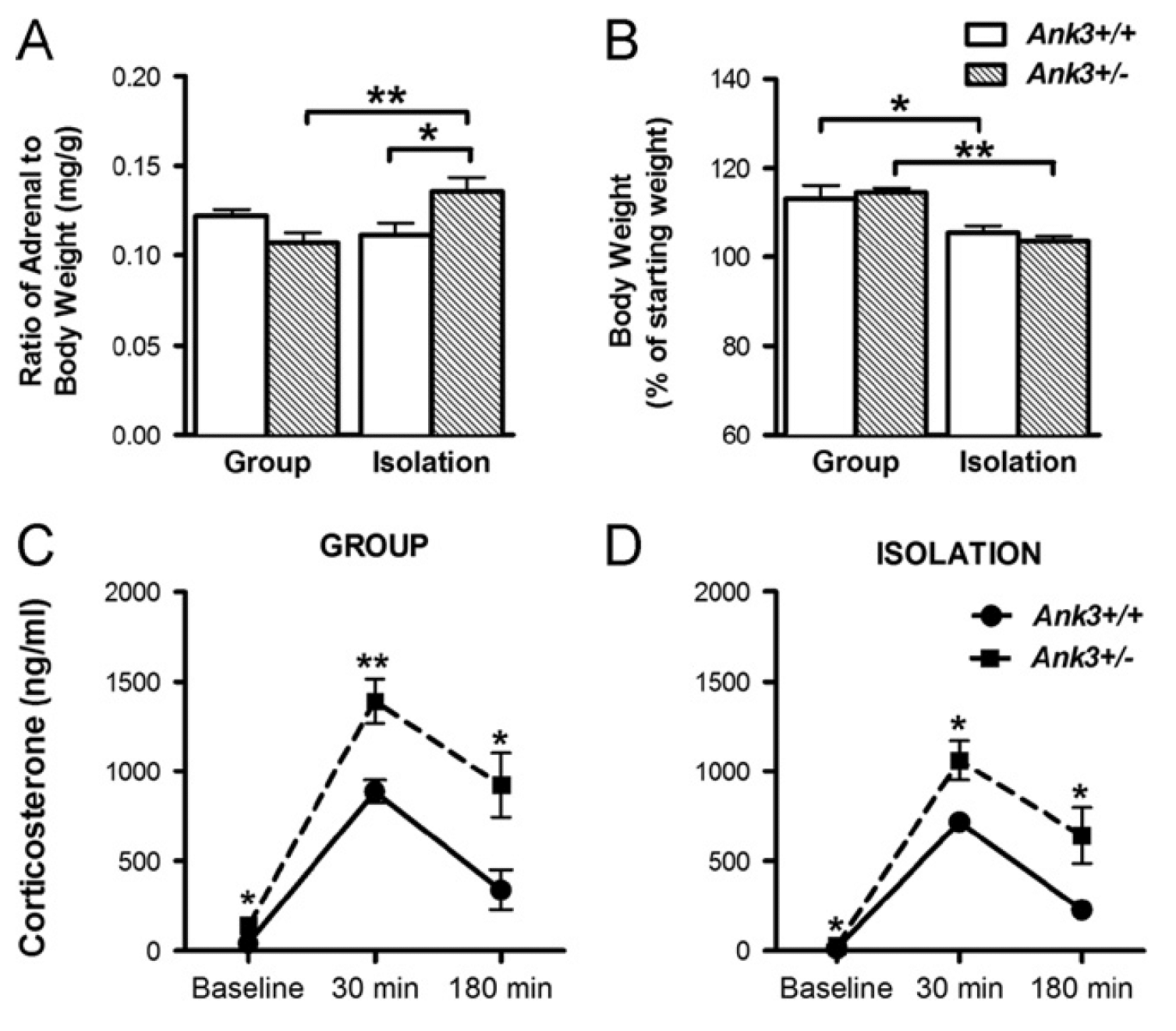
Ankyrin 3 (ANK3) has been strongly implicated as a risk gene for bipolar disorder (BD) by recent genome-wide association studies of patient populations. However, the genetic variants of ANK3 contributing to BD risk and their pathological function are unknown. This study defines a new role for Ank3 in the regulation of psychiatric-related behaviors and stress reactivity that lends support for its involvement in BD and establishes a general framework for determining the disease relevance of genes implicated by patient genome-wide association studies.
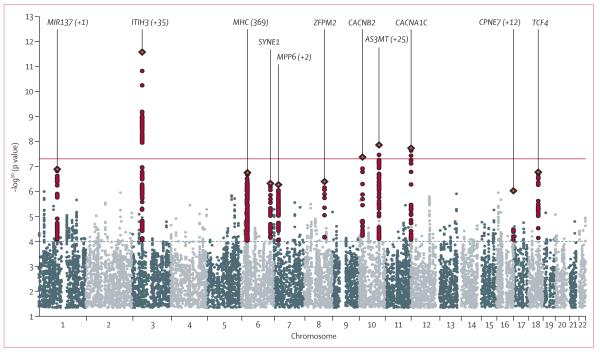
Findings from family and twin studies suggest that genetic contributions to psychiatric disorders do not in all cases map to present diagnostic categories. We aimed to identify specific variants underlying genetic effects shared between the five disorders in the Psychiatric Genomics Consortium: autism spectrum disorder, attention deficit-hyperactivity disorder, bipolar disorder, major depressive disorder, and schizophrenia. SNPs at four loci surpassed the cutoff for genome-wide significance (p<5x~10-8) in the primary analysis: regions on chromosomes 3p21 and 10q24, and SNPs within two L-type voltage-gated calcium channel subunits, CACNA1C and CACNB2. Model selection analysis supported effects of these loci for several disorders. Loci previously associated with bipolar disorder or schizophrenia had variable diagnostic specificity. Polygenic risk scores showed cross-disorder associations, notably between adult-onset disorders. Pathway analysis supported a role for calcium channel signaling genes for all five disorders. Finally, SNPs with evidence of cross-disorder association were enriched for brain eQTL markers.